
KEYTRUDA®(ペムブロリズマブ)とLENVIMA®(レンバチニブ)の併用療法について進行性子宮内膜がんに係る承認を米国 FDA より取得 免疫療法とチロシンキナーゼ阻害剤の併用療法による治療ラインに関わらず全身療法後に増悪した、根治的手術または放射線療法に不適応な高頻度マイクロサテライト不安定性を有さない、またはミスマッチ修復機構欠損を有さない進行性子宮内膜がんに関する承認 臨床試験では、全生存期間、無増悪生存期間および奏効率を統計学的に有意に改善し、進行性子宮内膜がんにおける切なるアンメット・ニーズに応えるものとなる
2021/07/30 00:00 JST
報道関係各位
MSD株式会社
この参考資料は、FDA Approves KEYTRUDA® (pembrolizumab) Plus LENVIMA® (lenvatinib) Combination for Patients With Certain Types of Advanced Endometrial Carcinoma の日本語訳であり、内容や解釈については英語が優先されます。適応症と安全性情報も米国のものであり、日本国内の情報ではありません。
KEYTRUDA®は、日本ではキイトルーダ®として、悪性黒色腫、切除不能な進行・再発の非小細胞肺癌、再発又は難治性の古典的ホジキンリンパ腫、がん化学療法後に増悪した根治切除不能な尿路上皮癌、がん化学療法後に増悪した進行・再発の高頻度マイクロサテライト不安定性(MSI-High)を有する固形癌(標準的な治療が困難な場合に限る)注)、根治切除不能又は転移性の腎細胞癌、再発又は遠隔転移を有する頭頸部癌、がん化学療法後に増悪したPD-L1陽性の根治切除不能な進行・再発の食道扁平上皮癌に対する効能又は効果で承認を取得しております。LENVIMA®は、日本ではレンビマ®として、根治切除不能な甲状腺癌、切除不能な肝細胞癌、切除不能な胸腺癌に対する効能又は効果で承認を取得しております。注) 条件付き早期承認対象
参考資料
KEYTRUDA®(ペムブロリズマブ)とLENVIMA®(レンバチニブ)の併用療法について
進行性子宮内膜がんに係る承認を米国 FDA より取得
免疫療法とチロシンキナーゼ阻害剤の併用療法による
治療ラインに関わらず全身療法後に増悪した、根治的手術または放射線療法に不適応な
高頻度マイクロサテライト不安定性を有さない、またはミスマッチ修復機構欠損を有さない
進行性子宮内膜がんに関する承認
臨床試験では、全生存期間、無増悪生存期間および奏効率を統計学的に有意に改善し、
進行性子宮内膜がんにおける切なるアンメット・ニーズに応えるものとなる
2021年7月22日 ニュージャージー州ケニルワース、ニュージャージー州ウッドクリフレイク―Merck & Co., Inc., Kenilworth, N.J., U.S.A.(米国とカナダ以外ではMSD)とエーザイ株式会社は、本日、Merck & Co., Inc., Kenilworth, N.J., U.S.A.の抗PD-1抗体KEYTRUDA®とエーザイ創製の経口チロシンキナーゼ阻害剤LENVIMA®の併用療法による、治療ラインに関わらず全身療法後に増悪した、根治的手術または放射線療法に不適応な高頻度マイクロサテライト不安定性(microsatellite instability-high: MSI-High)を有さない、またはミスマッチ修復機構欠損(mismatch repairdeficient: dMMR)を有さない進行性子宮内膜がんの適応について、米国食品医薬品局(FDA)より承認を取得したことをお知らせします。本患者集団に対する今回の承認は、第3相臨床試験 (KEYNOTE-775 試験/309 試験)の結果に基づいています。本試験において、対照薬の化学療法(治験医師選択によるドキソルビシンまたはパクリタキセル)と比較して、本併用療法は、全生存期間(Overall Survival: OS)を統計学的に有意に延長し、死亡リスクを32%減少させました(ハザード比(Hazard Ratio: HR)=0.68 [95% 信頼区間(Confidence Interval: CI),0.56-0.84]; p=0.0001)。また、無増悪生存期間(Progression-Free Survival: PFS)を統計学的に有意に延長し、増悪または死亡のリスクを 40%減少させました(HR=0.60 [95% CI, 0.50-0.72]; p<0.0001)。さらに、本併用療法の奏効率(Objective Response Rate: ORR)は 30%(95%CI: 26-36)であり、治験医師選択によるドキソルビシンまたはパクリタキセルの 15%(95%CI: 12-19)と比較して、統計学的に有意な改善を示しました。本併用療法と治験医師選択によるドキソルビシンまたはパクリタキセルの完全奏効率はそれぞれ 5%と 3%であり、部分奏効率はそれぞれ 25%と 13%でした。
KEYTRUDA®投与により、免疫関連有害事象は、あらゆる臓器または組織において発現する可能性があり、同時に複数の体組織に影響を及ぼす可能性や、重症または致死的なものが発現する可能性もあります。KEYTRUDA®投与中または投与後に発現する可能性のある免疫関連有害事象には、肺臓炎、大腸炎、肝炎、内分泌障害、腎機能障害、皮膚障害、実質臓器移植拒絶反応、および同種造血幹細胞移植合併症が含まれます。上記に記載されている免疫関連有害事象には、起こりうる重症および致死的な免疫関連有害事象のすべてが含まれていない可能性があります。KEYTRUDA®の安全な使用を担保する上で、免疫関連有害事象の早期の同定およびマネジメントは必要不可欠です。有害事象の重症度に基づき、必要に応じて、KEYTRUDA®を休薬または投与中止し、コルチコステロイドの投与が必要となります。KEYTRUDA®は重症または生命を脅かすinfusion reactionを引き起こす可能性があります。作用機序に基づき、KEYTRUDA®は、妊婦に投与されると胎児に悪影響を及ぼす可能性があります。
LENVIMA®投与により発現する可能性のある有害事象(Adverse reactions)には、高血圧、心機能障害、動脈血栓塞栓症、肝毒性、腎不全または腎機能障害、蛋白尿、下痢、瘻孔形成ならびに消化管穿孔、QT 間隔延長、低カルシウム血症、可逆性後白質脳症症候群、出血性イベント、甲状腺刺激ホルモン抑制障害/甲状腺機能障害、創傷治癒障害、および顎骨壊死が含まれ、重篤または致死的なものが発現する可能性があります。有害事象の種類および/またはその重症度により、LENVIMA®は休止、減量および/または、中止される可能性があります。作用メカニズムおよび動物での再現試験に基づき、LENVIMA®は、妊婦に投与されると致死的な出血を引き起こす可能性があります。妊娠の可能性のある女性は避妊が推奨されます。
本試験の治験責任医師であり、Memorial Sloan Kettering Cancer Center のメディカルオンコロジストである Vicky Makker 博士は、「根治的治療に不適応な進行性子宮内膜がんの 5 年生存率はわずか 17%であり、特に、全身化学療法後に増悪した患者さんの治療選択肢は限られています。本承認は、患者さんのこの治療困難な悪性腫瘍との闘いを支援する重要なステップであり、医師は、生存期間を延長する新たな治療選択肢を患者さんに提示することが可能となります」と述べています。
Merck & Co, Inc., Kenilworth, N.J., U.S.A. 研究開発本部 オンコロジークリニカルリサーチのバイスプレジデントである Gregory Lubiniecki 博士は、「本試験で使用された化学療法と比較して、本併用治療レジメンは、前治療歴のある進行性子宮内膜がん患者さんにおいて延命効果を示しました。今回の承認は、KEYTRUDA®とLENVIMA®の併用療法がすでに取得している進行性子宮内膜がんに対する迅速承認の検証試験としての第3相臨床試験の結果に基づくものであり、治療困難ながんに対する本併用療法の可能性を追求してきたエーザイとの共同研究の成果をさらに裏付けるものです」と述べています。
エーザイ株式会社の執行役 オンコロジービジネスグループ チーフメディスンクリエーションオフィサー兼チーフディスカバリーオフィサーである大和隆志博士は、「FDA による進行性子宮内膜がんにおける本併用療法の承認は、治療オプションが限られていた患者さんのコミュニティを救う重要なステップとなります。本承認は、我々が、がん患者さんのアンメット・ニーズへの対応を追求し続けてきた成果です。309 試験/KEYNOTE-775 試験に参加いただいた患者さんとそのご家族の皆様、および医療関係者の皆様に深く感謝します。本試験にご協力いただいた皆様のコミットメントのおかげで、我々は意義のあるマイルストンを達成できました」と述べています。
本併用療法は、FDA の迅速承認制度、リアルタイムオンコロジーレビュー(Real-Time OncologyReview:RTOR)パイロットプログラム、およびプロジェクトOrbis の先駆け制度の下、KEYNOTE-146 試験/111 試験の結果に基づき、全身療法後に増悪した、根治的手術または放射線療法に不適応な MSI-High を有さない、または dMMR を有さない進行性子宮内膜がんの適応で、既に承認を受けていました。迅速承認制度に従って、この迅速承認の継続には、臨床的有用性の検証と説明が要件となっていましたが、KEYNOTE-775 試験/309 試験の結果により、本要件は満たされ、今回の承認にいたりました。
本承認の基となった試験結果について
本承認は、ネオアジュバントおよびアジュバントを含むいずれかの治療ラインにおいて、少なくとも 1 レジメンのプラチナ製剤による前治療歴のある進行性子宮内膜がんを対象とした、多施設共同非盲検無作為化実薬対照の 827 人の患者を登録した第3相臨床試験(KEYNOTE-775試験/309 試験 ClinicalTrials.gov, NCT03517449)の結果に基づいています。癌肉腫を含む子宮肉腫の患者、活動性の自己免疫疾患または免疫抑制が必要な健康状態の患者は、本試験の登録に不適格と判断されました。MSI-High を有さない、または dMMR を有さない進行性子宮内膜がん患者は、Eastern Cooperative Oncology Group (ECOG) performance status (PS)(数値が小さいほど健康状態がよい)、地域、および骨盤照射による放射線治療歴によって層別化されました。患者は次の群に 1:1 で無作為に割り付けられました。
- KEYTRUDA®(200 mg、3週ごと静脈内投与)とLENVIMA®(20 mg、1日1 回経口投与)の併用
- 対照薬である治験医師選択によるドキソルビシン(60 mg/m2 3週ごと投与)またはパクリタキセル(80 mg/m2 週1回投与を3週連続し、1週間休薬)
治験薬の投与は、許容できない毒性、または RECIST v1.1(固形がんに対する腫瘍径の変化を効果判定に用いる評価基準)に基づく独立画像判定により増悪とされるまで継続されました。KEYTRUDA®の投与は最大で24カ月まで継続されました。本併用療法は、患者が臨床的に病勢安定と診断され、治験医師によって臨床的有用性および忍容性があると判断された場合、RECISTv1.1で定義された増悪後も投与継続が認められました。腫瘍評価は8週毎に行われました。主要な有効性評価項目は、OSおよびRECIST v1.1(標的病変の総数は最大10個、各臓器における最大数は5個に変更)に基づく独立画像判定によるPFSでした。その他の有効性評価項目には、独立画像判定によるORRおよび奏効期間が含まれました。
dMMR を有さない697人の患者のうち、346人がKEYTRUDA®とLENVIMA®の併用療法群に割り付けられ、351人が治験医師選択によるドキソルビシン(254人)またはパクリタキセル(97人)に無作為化されました。dMMRを有さない患者集団の特徴は、年齢の中央値65歳(範囲:30-86歳)、65歳以上52%、人種:白人62%、アジア人 22%、黒人3%、ECOG PS0 60%、ECOG PS1 40%でした。組織学的な分類は、類内膜癌55%、漿液性癌30%、明細胞癌7%、混合癌4%、その他3%でした。これら697人すべての患者に子宮内膜がんに対する全身療法治療歴があり、67%は1 回、30%は2 回、3%は3 回以上の全身療法治療歴がありました。37%の患者が術前補助療法または術後補助療法治療のみを受けていました。
MSI-High を有さない、または dMMR を有さない患者集団における有効性に関する試験結果は以下のとおりでした。
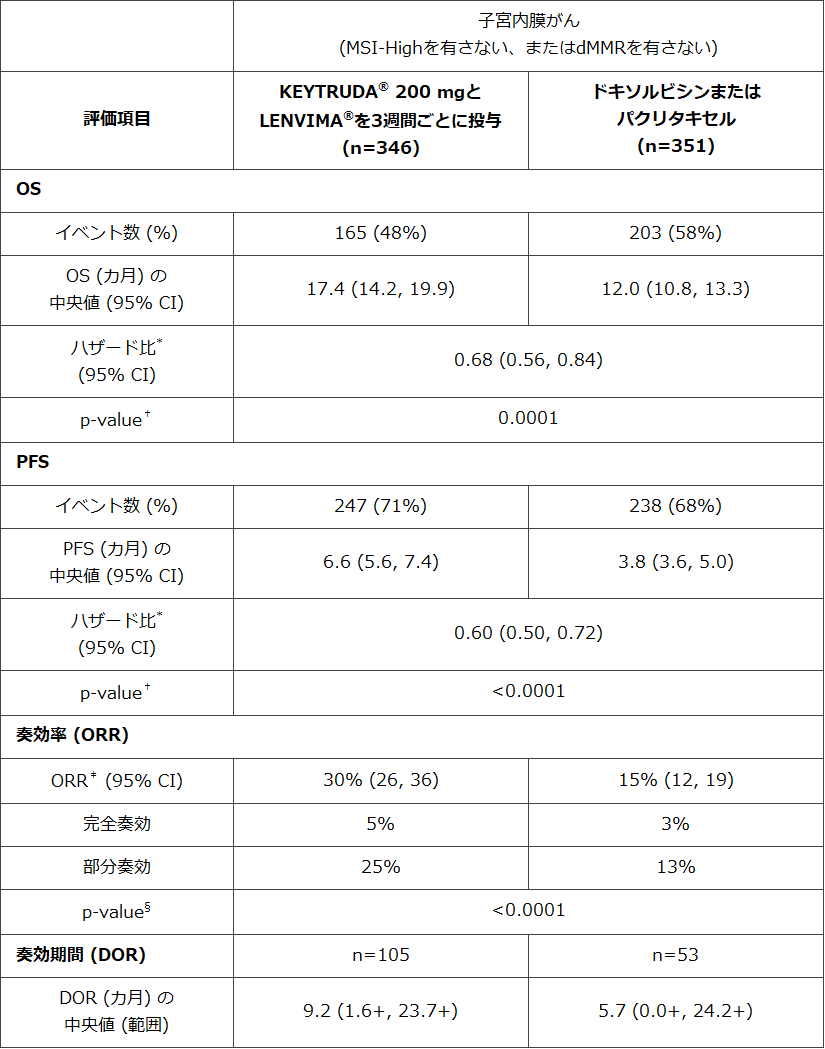
* 層別Cox回帰モデルに基づく
† 層別ログランク検定に基づく
‡ 奏効: 確定した完全奏効または部分奏効である最良総合効果
§ ECOG PS、地域、および骨盤照射による放射線治療歴によって層別化されたMiettinen and Nurminen法に基づく
腫瘍がMSI-Highを有さない、またはdMMRを有さない状態であった患者において、治験薬の投与期間の中央値は 7.2 カ月(範囲:1 日-26.8 カ月)でした。KEYTRUDA®の投与期間の中央値は 6.8 カ月(範囲:1 日-25.8 カ月)でした。LENVIMA®の投与期間の中央値は 6.7 カ月(範囲:1 日-26.8 カ月)でした。
致死的な有害事象はKEYTRUDA®とLENVIMA®の併用療法を受けた患者のうち、4.7%に発現し、2 例の肺炎、1 例の急性腎障害、急性心筋梗塞、大腸炎、食欲減退、腸管穿孔、下部消化管出血、悪性消化管閉塞、多臓器機能不全症候群、骨髄異形成症候群、肺塞栓症、および右室機能不全が含まれました。
KEYTRUDA®とLENVIMA®の併用療法を受けたこれらの患者のうち、50%に重篤な有害事象が発現しました。
発現した重篤な有害事象発現率 3%以上)は、高血圧(4.4%)および尿路感染(3.2%)でした。
KEYTRUDA®の投与中止に至った有害事象(adverse reaction、グレード 1-4)は、15%の患者で発現しました。LENVIMA®の投与中止に至った有害事象(adverse reaction、グレード 1-4)は、26%の患者で発現しました。KEYTRUDA®の投与中止に至った頻度の高い有害事象(発現率 1%以上)は、アラニンアミノトランスフェラーゼ(ALT)増加(1.2%)でした。LENVIMA®の投与中止に至った頻度の高い有害事象(発現率 1%以上)は、高血圧(2.0%)、無力症(1.8%)、下痢(1.2%)、食欲減退(1.2%)、蛋白尿(1.2%)、および嘔吐(1.2%)でした。
KEYTRUDA®の休薬に至った有害事象は 48%の患者に発現しました。LENVIMA®の休薬に至った有害事象は 58%の患者に発現しました。KEYTRUDA®の休薬に至った頻度の高い有害事象(発現率 3%以上)は、下痢(8%)、ALT 増加(4.4%)、AST 増加(3.8%)および高血(3.5%)でした。LENVIMA®の減量または休薬に至った頻度の高い有害事象(発現率 2%以上)は、高血圧(11%)、下痢(11%)、蛋白尿(6%)、食欲減退(5%)、嘔吐(5%)、ALT 増加(3.5%)、疲労(3.5%)、悪心(3.5%)、腹痛(2.9%)、体重減少(2.6%)、尿路感染(2.6%)、アスパラギン酸アミノトランスフェラーゼ(AST)増加(2.3%)、無力症(2.3%)、および手掌・足底発赤知覚不全症候群(2%)でした。
LENVIMA®投与を受けた患者の 67%は、有害事象によりLENVIMA®を減量されました。LENVIMA®の減量に至った頻度の高い有害事象(発現率 5%以上)は、高血圧(18%)、下痢(11%)、手掌・足底発赤知覚不全症候群(9%)、蛋白尿(7%)、疲労(7%)、食欲減退(6%)、無力症(5%)、および体重減少(5%)でした。
KEYTRUDA®とLENVIMA®の併用療法によって発現した頻度の高い有害事象(全グレード、発現率 20%以上)は、甲状腺機能低下症(67%)、高血圧(67%)、疲労(58%)、下痢(55%)、筋骨格系障害(53%)、悪心(49%)、食欲減退(44%)、嘔吐(37%)、口内炎(35%)、体重減少(34%)、腹痛(34%)、尿路感染(31%)、蛋白尿(29%)、便秘(27%)、頭痛(26%)、出血性イベント(25%)、手掌・足底発赤知覚不全症候群(23%)、発声障害(22%)、および発疹(20%)でした。
子宮内膜がんについて
子宮内膜がんは、子宮の内層に発生し、子宮における最も発生頻度の高いがんです。子宮体がんの罹患者数は 2020 年において、世界で 41 万 7 千人以上と推定され、約 9 万 7 千人以上が亡くなったとされています(これらの推定には子宮内膜がんに加えて子宮肉腫が含まれています。子宮内膜がんは子宮体がんの 90%以上を占めるとされていますが、子宮内膜がんのみの数はこの数よりもやや少ないと考えられます)。米国では 2021年に約 6 万 6 千人以上が新たに子宮体がんと診断され、約 1 万 3 千人が亡くなると推定されています。転移性子宮内膜がん(stage IV)の 5 年生存率は約 17%と推計されています。
KEYTRUDA®について
KEYTRUDA®は、自己の免疫力を高め、がん細胞を見つけて攻撃するのを助ける抗PD-1抗体です。KEYTRUDA®はPD-1とそのリガンドであるPD-L1およびPD-L2との相互作用を阻害して、がん細胞を攻撃する Tリンパ球を活性化するヒト化モノクローナル抗体です。
Merck & Co., Inc., Kenilworth, N.J., U.S.A.は業界最大のがん免疫療法臨床研究プログラムを行っており、現在1,500を超えるKEYTRUDA®の臨床試験を実施し、幅広い種類のがんや治療セッティングを検討しています。KEYTRUDA®の臨床プログラムでは、さまざまながんにおけるKEYTRUDA®の役割や、KEYTRUDA®による治療効果が得られる可能性を予測する因子について模索しており、さまざまなバイオマーカーの模索も行っています。
KEYTRUDA®用法・用量・安全性情報・Access Program・患者支援プログラムについて
用法・用量・安全性情報・Access Program・患者支援プログラムなど一部情報は米国のもので、日本の情報ではありません。詳しくは当社英文リリースをご参照ください。
LENVIMA®(レンバチニブ)カプセルついて
血管内皮増殖因子(VEGF)受容体であるVEGFR1(FLT1)、VEGFR2 (KDR)、VEGFR3 (FLT4)に対する選択的阻害活性を有する、経口投与可能なエーザイ創製のマルチキナーゼ阻害剤です。LENVIMA®は、線維芽細胞増殖因子(FGF)受容体のFGFR1-4、血小板由来増殖因子受容体(PDGFR)のPDGFRα、KIT、RETなどの腫瘍血管新生あるいは腫瘍悪性化に関与する受容体型チロシンキナーゼに対する選択的阻害活性を有します。マウスの腫瘍モデルにおいて、抗PD-1モノクローナル抗体併用により、LENVIMA®は、腫瘍関連マクロファージを減少させ、活性化細胞傷害性T細胞を増加させることで、それぞれの単剤療法を上回る抗腫瘍活性をもたらしました。
LENVIMA®安全性情報について
米国におけるLENVIMA®の安全性情報については、LENVIMA®製品ウェブサイト(http://www.lenvima.com )をご覧ください。
Merck & Co., Inc., Kenilworth, N.J., U.S.A.とエーザイによる戦略的提携について
2018年3月に、エーザイとMerck & Co., Inc., Kenilworth, N.J., U.S.A. (米国とカナダ以外ではMSD)は、LENVIMA®のグローバルな共同開発および共同販促を行う戦略的提携に合意しました。本合意に基づき、両社は、LENVIMA®について、単剤療法およびMerck & Co., Inc., Kenilworth, N.J., U.S.A.の抗PD-1抗体KEYTRUDA®の併用療法における共同開発、共同製造、共同販促を行います。
既に実施しているKEYTRUDA®とLENVIMA®の併用試験に加え、両社は新たにLEAP(LEnvatinib And Pembrolizumab)臨床プログラムを開始しました。これにより、KEYTRUDA®とLENVIMA®の併用療法は13種類のがんにおける20以上を超える臨床試験が進行中です。
Merck & Co., Inc., Kenilworth, N.J., U.S.A.のがん領域における取り組み
Merck & Co., Inc., Kenilworth, N.J., U.S.A.では、画期的な科学を革新的ながん治療薬に変換して世界中のがん患者さんを助けることに取り組んでいます。当社のオンコロジー事業にとって、がんと闘う人々を助けることは私たちの情熱であり、がん治療薬へアクセスしやすくすることは私たちの責任です。また、がん領域における取り組みの一環として、医薬品業界で一二を争う急成長を遂げている開発プログラムにより、30種類以上のがんに対するがん免疫療法の可能性を模索しています。また、引き続き戦略的買収を通じてポートフォリオを強化し、進行がんの治療を改善する可能性をもつ有望ながん治療薬候補の開発を最優先に進めています。当社のオンコロジー臨床試験について詳しくは、当社ウェブサイトをご覧ください。
Merck & Co., Inc., Kenilworth, N.J., U.S.A.について
Merck & Co., Inc., Kenilworth, N.J., U.S.A.(米国とカナダ以外ではMSD)は130年にわたり、人々の生命を救い、人生を健やかにするというミッションのもと、世界で最も治療が困難な病気のために、革新的な医薬品やワクチンの発見、開発、提供に挑みつづけてきました。また、多岐にわたる政策やプログラム、パートナーシップを通じて、患者さんの医療へのアクセスを推進する活動に積極的に取り組んでいます。私たちは、今日、がん、HIVやエボラといった感染症、そして新たな動物の疾病など、人類や動物を脅かしている病気の予防や治療のために、研究開発の最前線に立ち続け、世界最高の研究開発型バイオ医薬品企業を目指しています。詳細については、当社ウェブサイトやTwitter、Facebook、Instagram、YouTube、LinkedInをご参照ください。
エーザイのがん領域の取り組みについて
エーザイは、がん領域において、真の患者様ニーズが満たされておらず、かつ当社がフロントランナーとなり得る機会(立地)として、既存の創製薬による経験知を活かした「がん微小環境」とRNAスプライシングプラットフォーム等を用いた「ドライバー遺伝子変異とスプライシング異常」を標的とした抗がん剤の開発にフォーカスしています。これらの立地から新たな標的や作用機序を有する革新的新薬を創出し、がんの治癒の実現に向けて貢献することをめざしています。
エーザイについて
エーザイは、患者様とそのご家族の喜怒哀楽を第一義に考え、そのベネフィット向上に貢献する「ヒューマン・ヘルスケア(hhc)」を企業理念としています。当社はグローバルな研究開発・生産・販売拠点ネットワークを持ち、hhcの実現に向けて戦略的重要領域と位置づける「神経領域」「がん」を中心とするアンメット・メディカル・ニーズの高い疾患領域において、世界中の約1万人の社員が革新的な新薬の創出と提供に取り組んでいます。当社はhhcの理念のもと、サイエンス、臨床科学、患者様の視点から、顧みられない熱帯病、持続可能な開発目標(SDGs)を含む世界のアンメット・メディカル・ニーズに対して、革新的なソリューションの提供をめざします。
エーザイ株式会社の詳細情報は、www.eisai.com (グローバル), us.eisai.com (米国) またはhttp://www.eisai.eu (欧州、中東、アフリカ)をご覧ください。 Twitter (米国、グローバル) および LinkedIn (米国)でも情報公開しています。
Merck & Co., Inc., Kenilworth, N.J., U.S.A.の将来に関する記述
このニュースリリースには、米国の1995年私的証券訴訟改革法(the Private Securities Litigation Reform Act of 1995)の免責条項で定義された「将来に関する記述」が含まれています。これらの記述は、Merck & Co., Inc., Kenilworth, N.J., U.S.A.の経営陣の現時点での信条と期待に基づくもので、相当のリスクと不確実性が含まれています。新薬パイプラインに対する承認取得またはその製品化による収益を保証するものではありません。予測が正確性に欠けていた場合またはリスクもしくは不確実性が現実化した場合、実際の成果が、将来に関する記述で述べたものと異なる場合も生じます。
リスクと不確実性には、業界の一般的な状況および競争環境、金利および為替レートの変動などの一般的な経済要因、昨今の新型コロナウイルス感染症(COVID-19)の世界的大流行の影響、医薬品業界の規制やヘルスケア関連の米国法および国際法が及ぼす影響、ヘルスケア費用抑制の世界的な傾向、競合他社による技術的進歩や新製品開発および特許取得、承認申請などの新薬開発特有の問題、Merck & Co., Inc., Kenilworth, N.J., U.S.A.による将来の市況予測の正確性、製造上の問題または遅延、国際経済および政府の信用リスクなどの金融不安、画期的製品に対するMerck & Co., Inc., Kenilworth, N.J., U.S.A.の特許権やその他の保護の有効性への依存、特許訴訟や規制措置の対象となる可能性等がありますが、これらに限定されるものではありません。
Merck & Co., Inc., Kenilworth, N.J., U.S.A.は、新たな情報、新たな出来事、その他いかなる状況が加わった場合でも、将来に関する記述の更新を行う義務は負いません。将来に関する記述の記載と大きく異なる成果を招くおそれがあるこの他の要因については、Merck & Co., Inc., Kenilworth, N.J., U.S.A.に関するForm 10-Kの2020年度年次報告書および米国証券取引委員会(SEC)のインターネットサイト(www.sec.gov)で入手できるSECに対するその他の書類で確認できます。
# # #
MSDについて
MSD(Merck & Co., Inc., Kenilworth, N.J., U.S.A.が米国とカナダ以外の国と地域で事業を行う際に使用している名称)は、130年にわたり、人々の生命を救い、人生を健やかにするというミッションのもと、世界で最も治療が困難な病気のために、革新的な医薬品やワクチンの発見、開発、提供に挑みつづけてきました。MSDはまた、多岐にわたる政策やプログラム、パートナーシップを通じて、患者さんの医療へのアクセスを推進する活動に積極的に取り組んでいます。私たちは、今日、がん、HIVやエボラといった感染症、そして新たな動物の疾病など、人類や動物を脅かしている病気の予防や治療のために、研究開発の最前線に立ち続けています。MSDは世界最高の研究開発型バイオ医薬品企業を目指しています。MSDの詳細については、弊社ウェブサイト(www.msd.co.jp)やFacebook、Twitter、YouTubeをご参照ください。